Get Adjusted Matrix with scMerge2 parameter estimated
getAdjustedMat(
exprsMat,
fullalpha,
ctl = rownames(exprsMat),
adjusted_means = NULL,
ruvK = 20,
return_subset_genes = NULL
)Arguments
- exprsMat
A gene (row) by cell (column) matrix to be adjusted.
- fullalpha
A matrix indicates the estimated alpha returned by
scMerge2().- ctl
A character vector of negative control. It should have a non-empty intersection with the rows of exprsMat.
- adjusted_means
A rowwise mean of the gene by cell matrix
- ruvK
An integer indicates the number of unwanted variation factors that are removed, default is 20.
- return_subset_genes
An optional character vector of indicates the subset of genes will be adjusted.
Value
Returns the adjusted matrix will be return.
Examples
## Loading example data
data('example_sce', package = 'scMerge')
## Previously computed stably expressed genes
data('segList_ensemblGeneID', package = 'scMerge')
## Running an example data with minimal inputs
library(SingleCellExperiment)
#> Loading required package: SummarizedExperiment
#> Loading required package: MatrixGenerics
#> Loading required package: matrixStats
#>
#> Attaching package: ‘MatrixGenerics’
#> The following objects are masked from ‘package:matrixStats’:
#>
#> colAlls, colAnyNAs, colAnys, colAvgsPerRowSet, colCollapse,
#> colCounts, colCummaxs, colCummins, colCumprods, colCumsums,
#> colDiffs, colIQRDiffs, colIQRs, colLogSumExps, colMadDiffs,
#> colMads, colMaxs, colMeans2, colMedians, colMins, colOrderStats,
#> colProds, colQuantiles, colRanges, colRanks, colSdDiffs, colSds,
#> colSums2, colTabulates, colVarDiffs, colVars, colWeightedMads,
#> colWeightedMeans, colWeightedMedians, colWeightedSds,
#> colWeightedVars, rowAlls, rowAnyNAs, rowAnys, rowAvgsPerColSet,
#> rowCollapse, rowCounts, rowCummaxs, rowCummins, rowCumprods,
#> rowCumsums, rowDiffs, rowIQRDiffs, rowIQRs, rowLogSumExps,
#> rowMadDiffs, rowMads, rowMaxs, rowMeans2, rowMedians, rowMins,
#> rowOrderStats, rowProds, rowQuantiles, rowRanges, rowRanks,
#> rowSdDiffs, rowSds, rowSums2, rowTabulates, rowVarDiffs, rowVars,
#> rowWeightedMads, rowWeightedMeans, rowWeightedMedians,
#> rowWeightedSds, rowWeightedVars
#> Loading required package: GenomicRanges
#> Loading required package: stats4
#> Loading required package: BiocGenerics
#>
#> Attaching package: ‘BiocGenerics’
#> The following objects are masked from ‘package:stats’:
#>
#> IQR, mad, sd, var, xtabs
#> The following objects are masked from ‘package:base’:
#>
#> Filter, Find, Map, Position, Reduce, anyDuplicated, aperm, append,
#> as.data.frame, basename, cbind, colnames, dirname, do.call,
#> duplicated, eval, evalq, get, grep, grepl, intersect, is.unsorted,
#> lapply, mapply, match, mget, order, paste, pmax, pmax.int, pmin,
#> pmin.int, rank, rbind, rownames, sapply, setdiff, sort, table,
#> tapply, union, unique, unsplit, which.max, which.min
#> Loading required package: S4Vectors
#>
#> Attaching package: ‘S4Vectors’
#> The following object is masked from ‘package:utils’:
#>
#> findMatches
#> The following objects are masked from ‘package:base’:
#>
#> I, expand.grid, unname
#> Loading required package: IRanges
#> Loading required package: GenomeInfoDb
#> Loading required package: Biobase
#> Welcome to Bioconductor
#>
#> Vignettes contain introductory material; view with
#> 'browseVignettes()'. To cite Bioconductor, see
#> 'citation("Biobase")', and for packages 'citation("pkgname")'.
#>
#> Attaching package: ‘Biobase’
#> The following object is masked from ‘package:MatrixGenerics’:
#>
#> rowMedians
#> The following objects are masked from ‘package:matrixStats’:
#>
#> anyMissing, rowMedians
scMerge2_res <- scMerge2(exprsMat = logcounts(example_sce),
batch = example_sce$batch,
ctl = segList_ensemblGeneID$mouse$mouse_scSEG,
return_matrix = FALSE)
#> [1] "Cluster within batch"
#> Warning: You're computing too large a percentage of total singular values, use a standard svd instead.
#> Warning: You're computing too large a percentage of total singular values, use a standard svd instead.
#> [1] "Normalising data"
#> [1] "Constructing pseudo-bulk"
#> Dimension of pseudo-bulk expression: [1] 1047 131
#> [1] "Identifying MNC using pseudo-bulk:"
 #> [1] "Running RUV"
cosineNorm_mat <- batchelor::cosineNorm(logcounts(example_sce))
adjusted_means <- DelayedMatrixStats::rowMeans2(cosineNorm_mat)
newY <- getAdjustedMat(cosineNorm_mat, scMerge2_res$fullalpha,
ctl = segList_ensemblGeneID$mouse$mouse_scSEG,
ruvK = 20,
adjusted_means = adjusted_means)
assay(example_sce, "scMerge2") <- newY
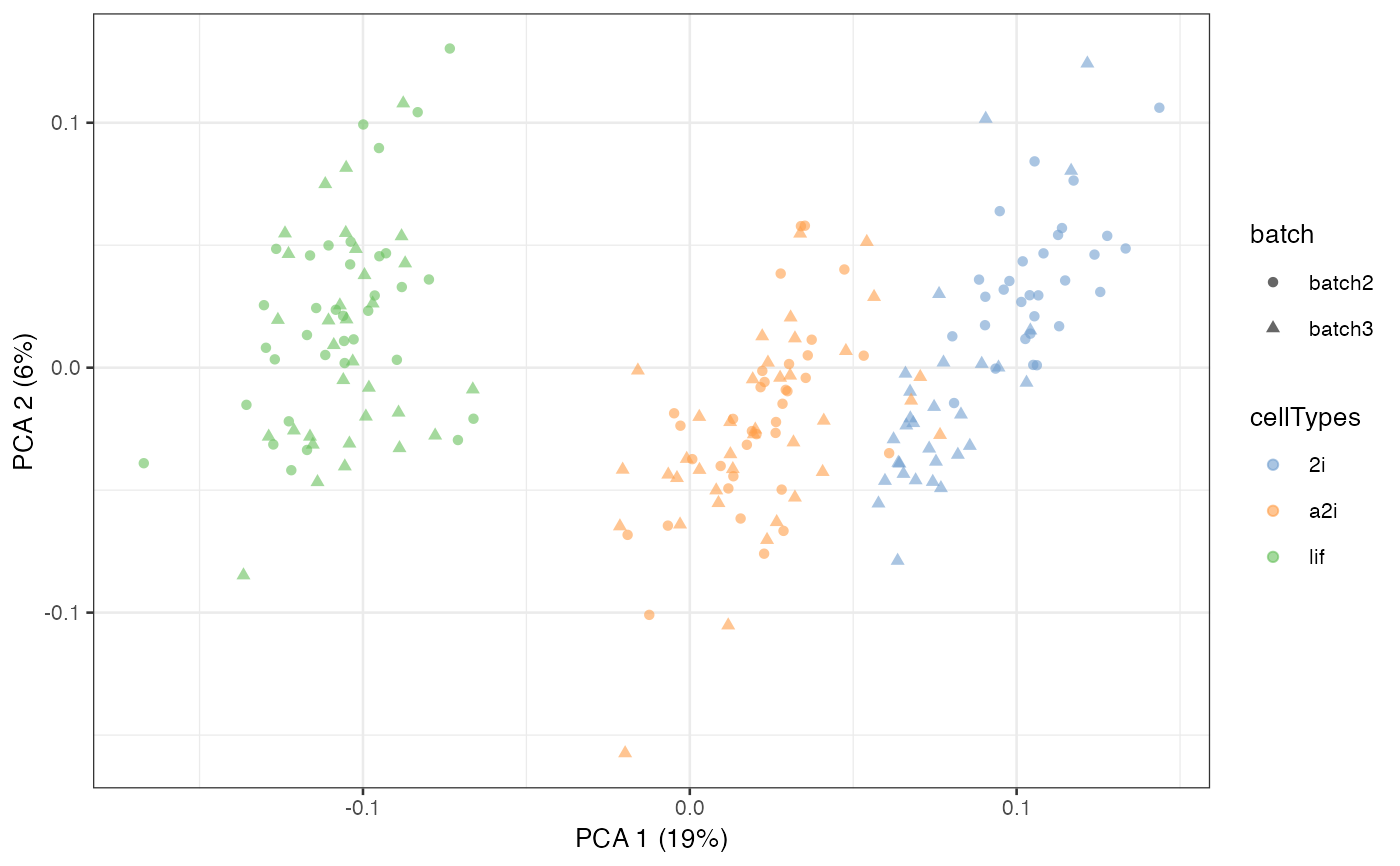
example_sce = scater::runPCA(example_sce, exprs_values = 'scMerge2')
scater::plotPCA(example_sce, colour_by = 'cellTypes', shape = 'batch')
#> [1] "Running RUV"
cosineNorm_mat <- batchelor::cosineNorm(logcounts(example_sce))
adjusted_means <- DelayedMatrixStats::rowMeans2(cosineNorm_mat)
newY <- getAdjustedMat(cosineNorm_mat, scMerge2_res$fullalpha,
ctl = segList_ensemblGeneID$mouse$mouse_scSEG,
ruvK = 20,
adjusted_means = adjusted_means)
assay(example_sce, "scMerge2") <- newY
example_sce = scater::runPCA(example_sce, exprs_values = 'scMerge2')
scater::plotPCA(example_sce, colour_by = 'cellTypes', shape = 'batch')