Merge single-cell data hierarchically from different batches and experiments leveraging (pseudo)-replicates, control genes and pseudo-bulk.
scMerge2h(
exprsMat,
batch_list = list(),
h_idx_list = list(),
cellTypes = NULL,
condition = NULL,
ctl = rownames(exprsMat),
chosen.hvg = NULL,
ruvK_list = 20,
use_bpparam = BiocParallel::SerialParam(),
use_bsparam = BiocSingular::RandomParam(),
use_bnparam = BiocNeighbors::AnnoyParam(),
pseudoBulk_fn = "create_pseudoBulk",
k_pseudoBulk = 30,
k_celltype = 10,
exprsMat_counts = NULL,
cosineNorm = TRUE,
return_subset = FALSE,
return_subset_genes = NULL,
return_matrix = TRUE,
verbose = TRUE,
seed = 1
)Arguments
- exprsMat
A gene (row) by cell (column) log-transformed matrix to be adjusted.
- batch_list
A list indicating the batch information for each cell in the batch-combined matrix.
- h_idx_list
A list indicating the indeces information in the hierarchical merging.
- cellTypes
An optional vector indicating the cell type information for each cell in the batch-combined matrix. If it is
NULL, pseudo-replicate procedure will be run to identify cell type.- condition
An optional vector indicating the condition information for each cell in the batch-combined matrix.
- ctl
A character vector of negative control. It should have a non-empty intersection with the rows of exprsMat
- chosen.hvg
An optional character vector of highly variable genes chosen.
- ruvK_list
An integer indicates the number of unwanted variation factors that are removed, default is 20.
- use_bpparam
A
BiocParallelParamclass object from theBiocParallelpackage is used. Default is SerialParam().- use_bsparam
A
BiocSingularParamclass object from theBiocSingularpackage is used. Default is RandomParam().- use_bnparam
A
BiocNeighborsParamclass object from theBiocNeighborspackage is used. Default is AnnoyParam().- pseudoBulk_fn
A character indicates the way of pseudobulk constructed.
- k_pseudoBulk
An integer indicates the number of pseudobulk constructed within each cell grouping. Default is 30.
- k_celltype
An integer indicates the number of nearest neighbours used in
buildSNNGraphwhen grouping cells within each batch. Default is 10.- exprsMat_counts
A gene (row) by cell (column) counts matrix to be adjusted.
- cosineNorm
A logical vector indicates whether cosine normalisation is performed on input data.
- return_subset
If
TRUE, adjusted matrix of only a subset of genes (hvg or indicates inreturn_subset_genes) will be return.- return_subset_genes
An optional character vector of indicates the subset of genes will be adjusted.
- return_matrix
A logical vector indicates whether the adjusted matrix is calculated and returned. If
FALSE, then only the estimated parameters will be returned.- verbose
If
TRUE, then all intermediate steps will be shown. Default toFALSE.- seed
A numeric input indicates the seed used.
Examples
## Loading example data
data('example_sce', package = 'scMerge')
## Previously computed stably expressed genes
data('segList_ensemblGeneID', package = 'scMerge')
# Create a fake sample information
example_sce$sample <- rep(c(1:4), each = 50)
# Construct a hierarchical index list
h_idx_list <- list(level1 = split(1:200, example_sce$batch),
level2 = list(1:200))
# Construct a batch information list
batch_list <- list(level1 = split(example_sce$sample, example_sce$batch),
level2 = list(example_sce$batch))
library(SingleCellExperiment)
exprsMat <- scMerge2h(exprsMat = logcounts(example_sce),
batch_list = batch_list,
h_idx_list = h_idx_list,
ctl = segList_ensemblGeneID$mouse$mouse_scSEG,
ruvK_list = c(2, 5))
#> [1] "Hierarchical merging level 1, data1"
#> [1] "Cluster within batch"
#> Warning: more singular values/vectors requested than available
#> Warning: You're computing too large a percentage of total singular values, use a standard svd instead.
#> Warning: requested number of components greater than available rank
#> Warning: more singular values/vectors requested than available
#> Warning: You're computing too large a percentage of total singular values, use a standard svd instead.
#> Warning: requested number of components greater than available rank
#> [1] "Normalising data"
#> [1] "Constructing pseudo-bulk"
#> Dimension of pseudo-bulk expression: [1] 1047 89
#> [1] "Identifying MNC using pseudo-bulk:"
#> [1] "Running RUV"
#> [1] "Hierarchical merging level 1, data2"
#> [1] "Cluster within batch"
#> Warning: more singular values/vectors requested than available
#> Warning: You're computing too large a percentage of total singular values, use a standard svd instead.
#> Warning: requested number of components greater than available rank
#> Warning: more singular values/vectors requested than available
#> Warning: You're computing too large a percentage of total singular values, use a standard svd instead.
#> Warning: requested number of components greater than available rank
 #> [1] "Normalising data"
#> [1] "Constructing pseudo-bulk"
#> Dimension of pseudo-bulk expression: [1] 1047 86
#> [1] "Identifying MNC using pseudo-bulk:"
#> [1] "Normalising data"
#> [1] "Constructing pseudo-bulk"
#> Dimension of pseudo-bulk expression: [1] 1047 86
#> [1] "Identifying MNC using pseudo-bulk:"
 #> [1] "Running RUV"
#> [1] "Hierarchical merging level 2, data1"
#> [1] "Cluster within batch"
#> [1] "Normalising data"
#> [1] "Constructing pseudo-bulk"
#> Dimension of pseudo-bulk expression: [1] 1047 100
#> [1] "Identifying MNC using pseudo-bulk:"
#> [1] "Running RUV"
#> [1] "Hierarchical merging level 2, data1"
#> [1] "Cluster within batch"
#> [1] "Normalising data"
#> [1] "Constructing pseudo-bulk"
#> Dimension of pseudo-bulk expression: [1] 1047 100
#> [1] "Identifying MNC using pseudo-bulk:"
 #> [1] "Running RUV"
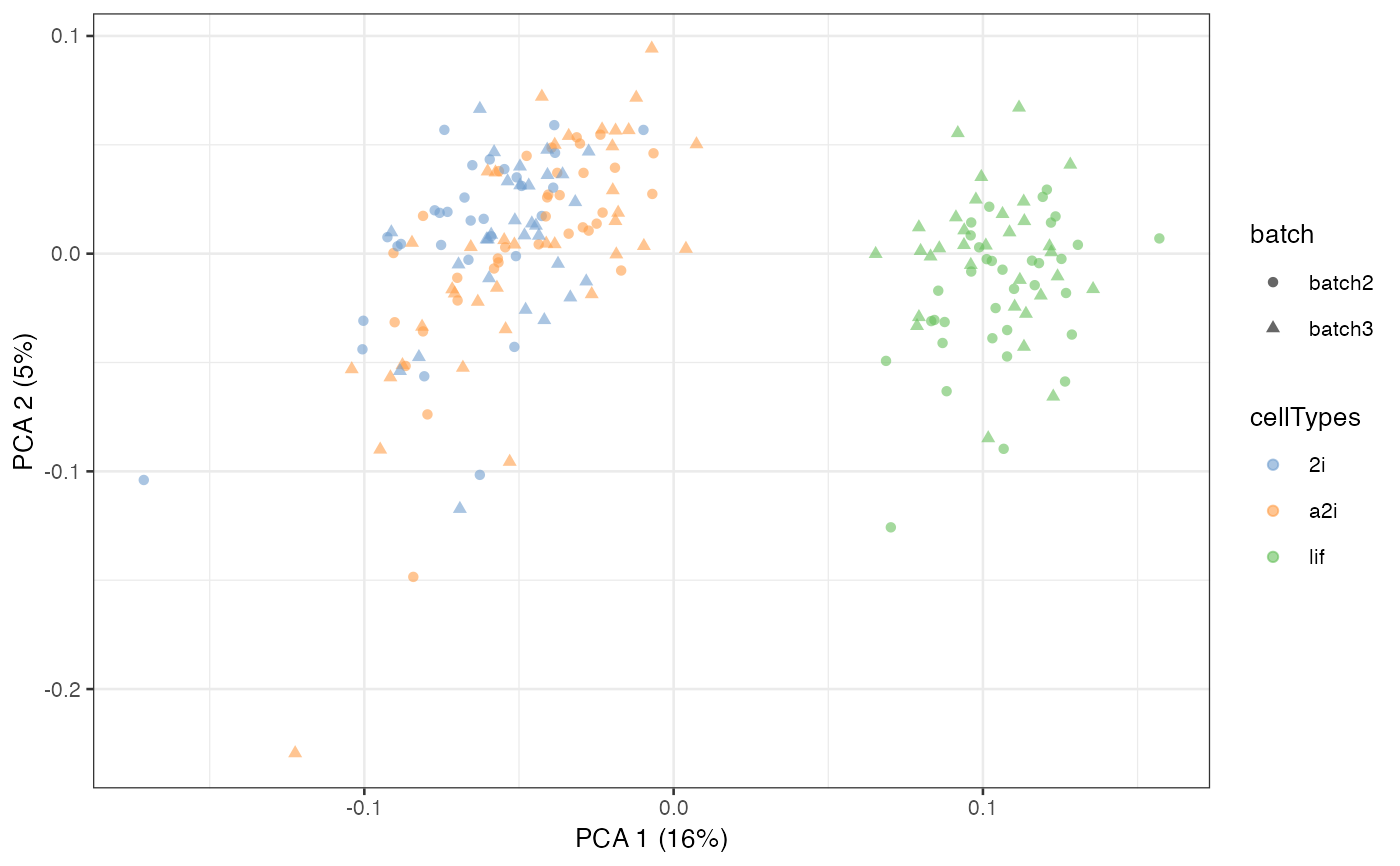
assay(example_sce, "scMerge2") <- exprsMat[[length(h_idx_list)]]
example_sce = scater::runPCA(example_sce, exprs_values = 'scMerge2')
scater::plotPCA(example_sce, colour_by = 'cellTypes', shape = 'batch')
#> [1] "Running RUV"
assay(example_sce, "scMerge2") <- exprsMat[[length(h_idx_list)]]
example_sce = scater::runPCA(example_sce, exprs_values = 'scMerge2')
scater::plotPCA(example_sce, colour_by = 'cellTypes', shape = 'batch')