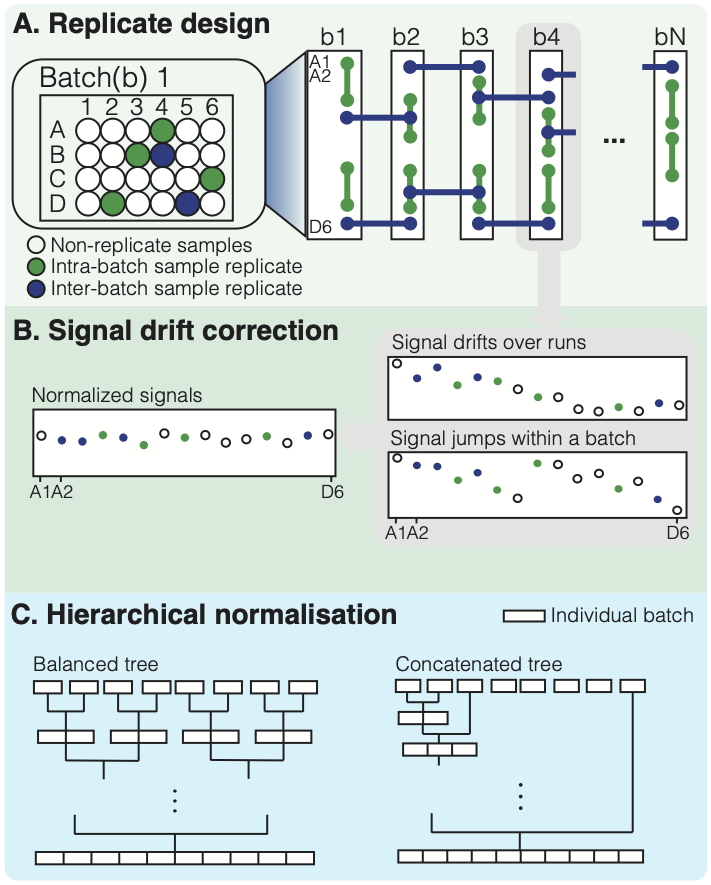
hRUV is a package for normalisation of multiple batches of metabolomics data in a hierarchical strategy with use of samples replicates in large-scale studies. The tool utilises 2 types of replicates: intra-batch and inter-batch replicates to estimate the unwatned variation within and between batches with RUV-III. We have designed the replicate embedding arrangements within and between batches from http://shiny.maths.usyd.edu.au/hRUV/. Our novel tool is a novel hierarchical approach to removing unwanted variation by harnessing information from sample replicates embedded in the seequence of experimental runs/batches and applying signal drift correction with robust linear or non-linear smoothers.
Software requirements
This package has been tested for macOS Big Sur (11.1) and Linux Debian 10 (buster) with R version 4.0.3.
Installation
Install the R package from GitHub using the devtools package:
if (!("devtools" %in% rownames(installed.packages())))
install.packages("devtools")
# Tested on devtools version 2.3.2.
library(devtools)
devtools::install_github("SydneyBioX/hRUV", build_vignettes=TRUE, dependencies = TRUE)Vignette
The vignette is available at https://sydneybiox.github.io/hRUV/. Alternatively, it can be accessed with browseVignettes("hRUV") in your R console.
Contact us
If you have any enquiries, especially about performing hRUV to integrate your metabolomics data, please contact taiyun.kim@sydney.edu.au. We are also happy to receive any suggestions and comments.
Citation
Kim, T., Tang, O., Vernon, S. T., Kott, K. A., Koay, Y. C., Park, J., James, D. E., Grieve, S. M., Speed, T. P., Yang, P., Figtree, G. A., O’Sullivan, J. F., & Yang, J. Y. H. (2021). A hierarchical approach to removal of unwanted variation for large-scale metabolomics data. In Nature Communications (Vol. 12, Issue 1). Springer Science and Business Media LLC. https://doi.org/10.1038/s41467-021-25210-5