library(ClassifyR)2 Procedure 1: Comparing cross-validated results
2.1 Introduction
Procedure 1 aims to compare the sample accuracy of breast cancer subtype classification using bulk gene expression data and histologically predicted gene expression data. Sample accuracy is defined as the proportion of correctly predicted classifications over multiple repeats. For example, if 70 out of 100 repeats correctly classify an individual, their sample accuracy is 0.70.
This analysis involves 54 oestrogen receptor-positive (ER+) and progesterone receptor-positive (PR+) breast cancer patients, with each assay containing expression values for 268 genes. The classification outcomes are ‘Subtype 1’ and ‘Subtype 2’, found by unsupervised machine learning. Subtype 1 corresponds to patients with higher expression of LPL, CAVIN2, and TIMP4 in macrophage cells, and ADIPOQ in stromal cells, associated with better survival. Subtype 2 is the opposite, associated with poorer survival. More details on the subtypes can be found at https://www.biorxiv.org/content/10.1101/2024.07.02.601790v1.full.
2.2 Setting up the environment and data objects
1. Load the R packages into the R environment
Timing ~ 6.5s
ClassifyR is used to perform all the demonstrated analyses.
2. Import preprocessed datasets for analysis
Timing ~ 0.04s
ghistMAE <- readRDS("data/procedure1/ghist_multiassayexperiment.rds")This command reads in a MultiAssayExperiment with two assays. The first is bulk gene expression data for 54 breast cancer individuals and the second is bulk gene expression as predicted from histological images of the same individuals. They are respectively named gene_expression and histology_inferred.
2.3 Cross-validated Classification
3. Classifying patients into subtype outcomes
Timing ~ 45.0s
set.seed(1)
classifyResult <- crossValidate(ghistMAE, outcome = "subtype", nFolds = 5, nRepeats = 100, nCores = 8)The set.seed(1) command ensures that any subsequent operations involving randomness yield consistent results across runs.
The next command uses the crossValidate function to perform 5-fold cross-validation with the automatically selected RandomForest classifier on both datasets. This process is repeated 100 times and utilizes 5 CPU cores for parallel processing to speed up classification. The type of classifier, number of folds, repeats and cores used can be adjusted as wished for different analyses. The outcome here is “subtype”, a column from colData(ghist_mae) containing the two breast cancer subtypes to be predicted.
2.4 Classification Evaluation
4. Visualising the classification performance
Timing ~ 1.75s
performancePlot(classifyResult)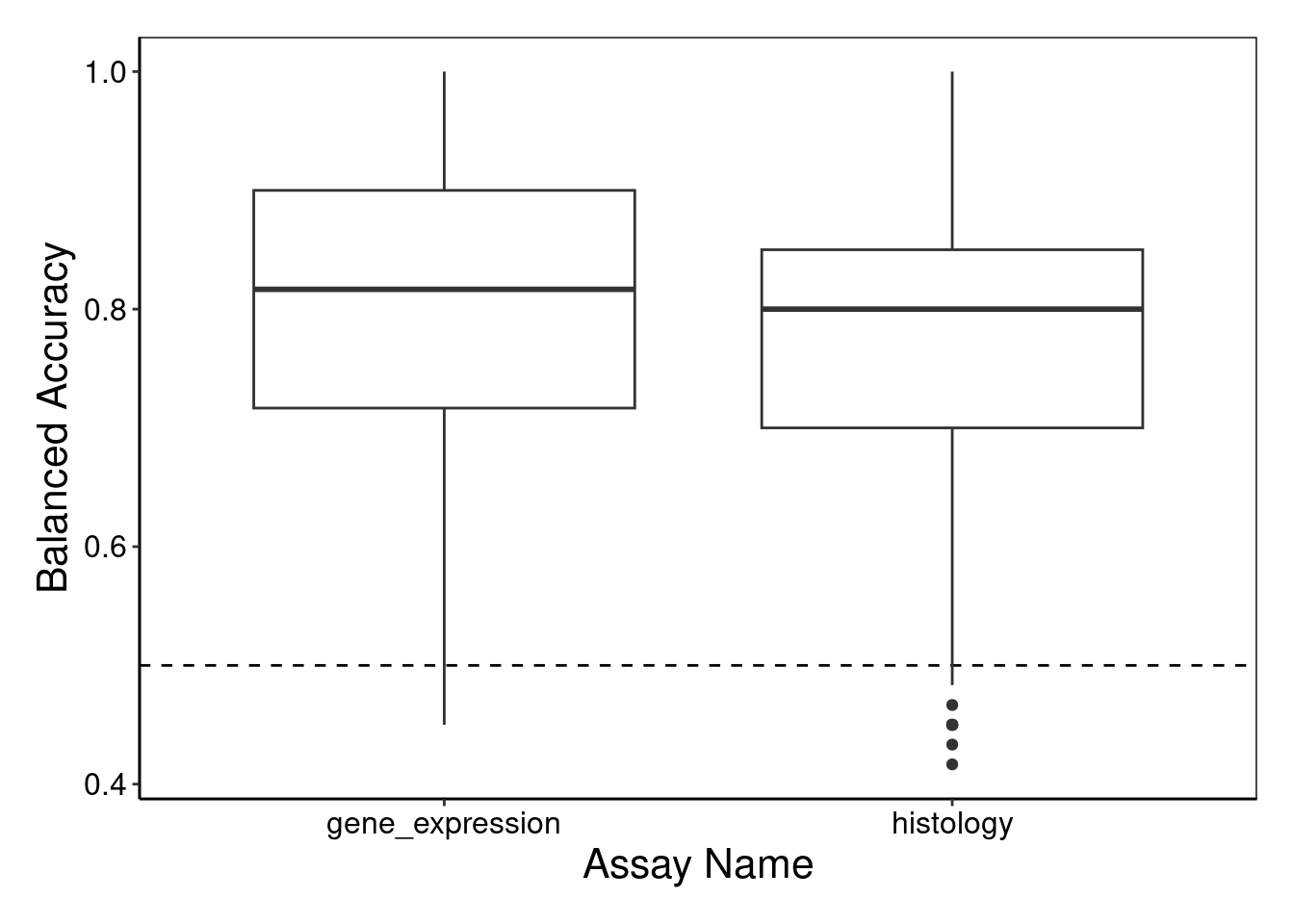
png
2 performancePlot outputs a side-by-side boxplot of the balanced accuracies for each dataset.
Both methods perform comparably in terms of median balanced accuracy and also demonstrate similar distributions of performance as seen from the comparable interquartile range and range. There does not appear to be much difference in the classification performance of both assays.
samplesMetricMap(classifyResult, showXtickLabels = FALSE)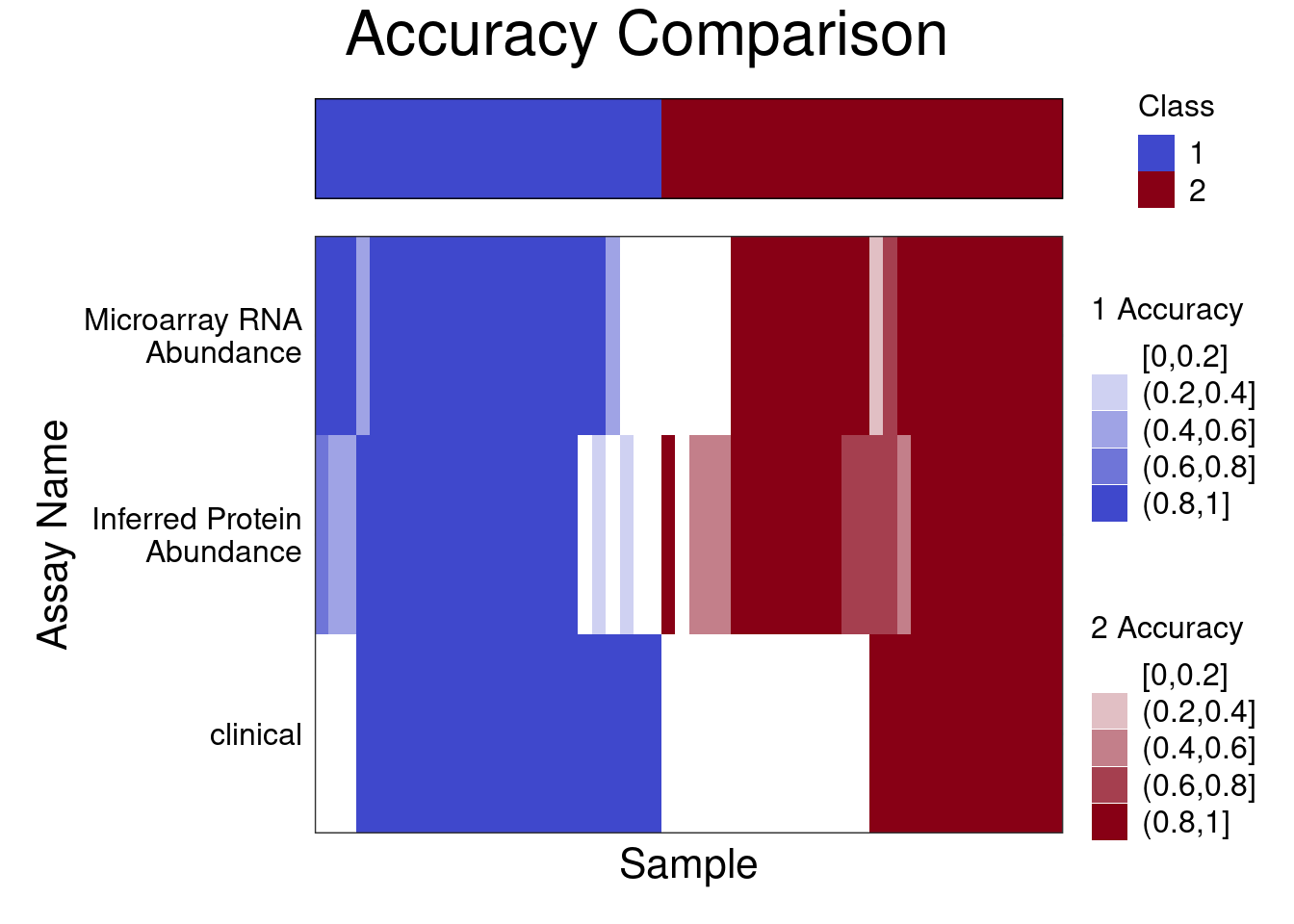
TableGrob (2 x 1) "arrange": 2 grobs
z cells name grob
1 1 (2-2,1-1) arrange gtable[layout]
2 2 (1-1,1-1) arrange text[GRID.text.550]png
2 samplesMetricMap outputs a heatmap showing the classification accuracy for each of 100 repeats in each sample. A greater proportion of samples show high sample accuracies (0.8,1] when classified by the expression data as opposed to the histological data.