library(readr)
library(tidyverse)
library(ClassifyR)
# Load the PROMAD data
promad <- readRDS("data/procedure4/promad.rds")5 Procedure 4
The transferability of biomarkers from one patient population to another is often difficult to fully capture. Here we present two complementary approaches to assess and build transferable models from gene expression data: a pairwise cross-validation method (criss-cross validation) and an optional leave-one-dataset-out framework using Transferable Omics Prediction (TOP).
5.1 Intro
Procedure 4 demonstrates the transferability of biomarkers from one patient population to another using a pairwise cross-validation approach, which we refer to as criss-cross validation. This is a common challenge in bioinformatics, where a model built on one cohort may not generalize well to others due to differences in population structure, data-generation protocols, or confounding variables.
Here, we introduce the function crissCrossValidate, which implements this pairwise validation for multiple datasets. In addition, we optionally demonstrate the Transferable Omics Prediction (TOP) approach (if you set runTOP = TRUE) for a leave-one-dataset-out framework that can further improve biomarker robustness. In this procedure, we use a simplified example dataset from the PROMAD atlas, which contains gene expression data from different organ transplant populations.
5.2 Setting up the data
What is in promad: - measurements: A named list of matrices (or data frames), where each element corresponds to one cohort’s gene expression data. Each row represents a patient, and each column represents a gene. - outcome: A named list of outcome vectors (e.g., factors for classification). Each element corresponds to one cohort, and the order of the outcomes should match the order of the gene expression data.
5.2.1 Pairwise cross-validation with crissCrossValidate
Criss-cross validation is an approach where each dataset is used to train a model (or select features) while all other datasets are used for testing. This results in a matrix of performance measures, giving a quick glimpse at how well biomarkers transfer among multiple cohorts.
set.seed(51773)
CCV <- crissCrossValidate(measurements = promad$measurements, outcomes = promad$outcome,
classifier = "LASSOGLM", performanceType = "AUC", runTOP = TRUE,
nFeatures = 100, nFolds = 5, nRepeats = 10,
nCores = 8)
crissCrossPlot(CCV, includeValues = TRUE, showResubMetric = FALSE)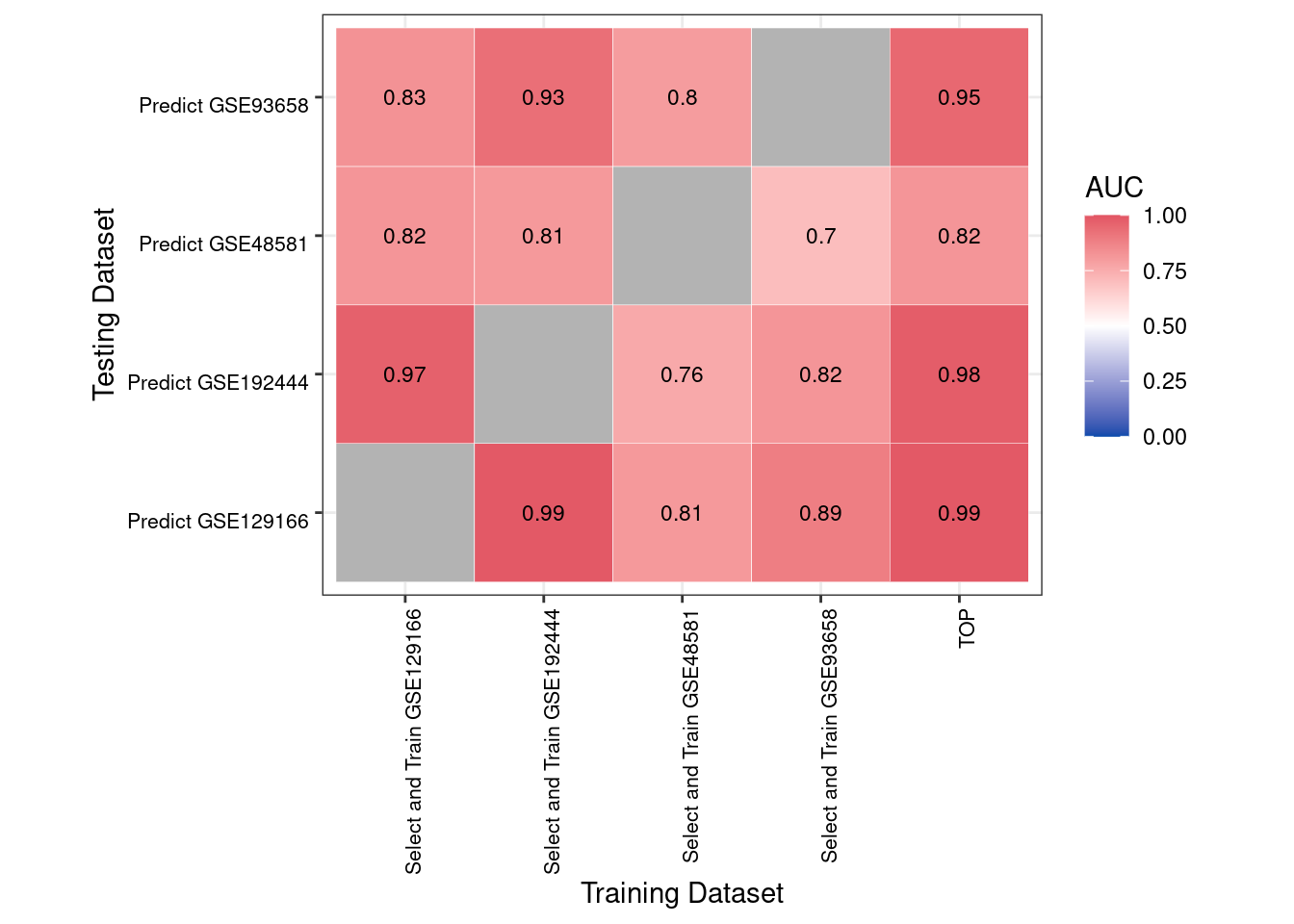
png
2 Interpretation of Results 1. Heatmap Rows: The dataset (cohort) from which features are selected or the model is trained. 2. Heatmap Columns: The dataset on which those selected features or model are tested. 3. Diagonal Cells: When showResubMetric = FALSE, the diagonal cells are set to NA (and may appear grayed out). Enabling showResubMetric = TRUE reveals self-prediction (resubstitution) performance, which typically inflates accuracy relative to true external validation.